Tutorial 6 - Soprano for defect calculations#
_
/|_|\
/ / \ \
/_/ \_\
\ \ / /
\ \_/ /
\|_|/
SOPRANO: a Python library for generation, manipulation and analysis of large batches of crystalline structures
Developed within the CCP-NC project. Copyright STFC 2022
# Basic imports
import os, sys
sys.path.insert(0, os.path.abspath('..')) # This to add the Soprano path to the PYTHONPATH
# so we can load it without installing it
# Other useful imports
import numpy as np
import ase
from ase import Atoms
from ase.build import bulk, molecule
from weas_widget import WeasWidget
%matplotlib inline
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
1 - Creating defect structures#
Soprano has a number of functionalities dedicated to defect calculations. Some of these have been used in work on finding the site in which a muonium pseudo-atom would sit after relaxing inside a crystal ( L. Liborio, S. Sturniolo et al, Computational prediction of muon stopping sites using ab initio random structure searching (AIRSS) ).
First, Soprano provides a number of generator functions to create defect structures, either randomly or in a specific way.
defectGen allows creating structures with a single added interstitial defect, distributed in a way to fill the empty space in between atoms while never creating any two configurations where the defects are closer than a given radius, using a so-called Poisson sphere distribution. These are good starting points for a random structure search approach.
from soprano.collection import AtomsCollection
from soprano.collection.generate import defectGen, substitutionGen, additionGen
si2 = bulk('Si') # Bulk silicon
dG = defectGen(si2, 'H', poisson_r=0.6, vdw_scale=0.5) # poisson_r controls the minimum distance between two defects,
# vdw_scale the one between defects and existing atoms
# (proportional to both's Van der Waals radius)
dColl = AtomsCollection(dG)
# The smaller poisson_r, the more structures are generated. The process is random and the final number isn't fixed.
print('{0} structures generated'.format(len(dColl)))
101 structures generated
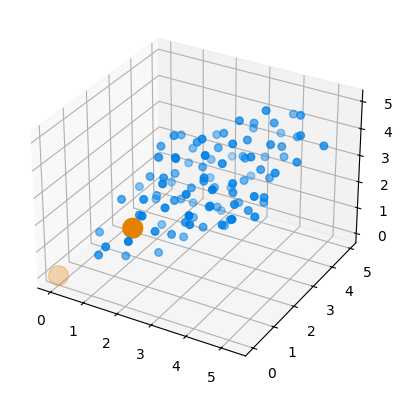
# By default, the defect is placed in position 0 in each new structure
def_pos = dColl.all.get_positions()[:,0]
fig = plt.figure()
ax = fig.add_subplot(111, projection='3d')
ax.scatter3D(*def_pos.T, color=(0, 0.5, 0.9), s=30) # Hydrogen defects are blue
ax.scatter3D(*si2.get_positions().T, color=(0.9,0.5,0), s=200) # Silicon atoms are orange
plt.show()
# Interactive viewer: show the Si+H defect structures
# The defect H is always placed at index 0 in each structure
from weas_widget import WeasWidget
v = WeasWidget()
v.from_ase(dColl.structures)
v.avr.model_style = 1
v.avr.color_type = "VESTA"
v.avr.boundary = [[-0.1, 1.1], [-0.1, 1.1], [-0.1, 1.1]]
v.avr.show_bonded_atoms = True
v.avr.highlight.settings['defect'] = {
"indices": [0],
"type": "sphere",
"color": [0.2, 0.5, 0.9],
"scale": 1.5,
"opacity": 0.5,
}
v
substitutionGen allows to simply replace one or more existing atoms with new ones. The replacements can be limited to only a specific selection, and can be accepted or rejected with a test function.
from soprano.properties.linkage import Bonds
from soprano.selection import AtomSelection
# As an example, we generate a molecule of ethyl mercaptan
ethmerc = molecule('CH3CH2SH')
syms = ethmerc.get_chemical_symbols()
bonds, bondmat = Bonds(return_matrix=True)(ethmerc)
# Where are the hydrogens? And the sulfur?
hsel = AtomSelection.from_element(ethmerc, 'H')
ssel = AtomSelection.from_element(ethmerc, 'S')
def isnot_S_bonded(s, subs):
# Return True only if none of the atoms in subs is bonded to sulfur
return np.all(bondmat[ssel.indices, list(subs)] == 0)
# Substitute two hydrogens at a time with chlorine, but only if neither is bonded to sulfur
sG = substitutionGen(ethmerc, 'Cl', to_replace=hsel, n=2, accept=isnot_S_bonded)
sColl = AtomsCollection(sG)
print('{0} structures generated'.format(len(sColl)))
10 structures generated
WARNING: divide by zero encountered in divide (/home/runner/work/soprano/soprano/soprano/utils.py, line: 354)
WARNING: invalid value encountered in dot (/home/runner/work/soprano/soprano/soprano/utils.py, line: 354)
WARNING: invalid value encountered in cast (/home/runner/work/soprano/soprano/soprano/utils.py, line: 361)
# Interactive viewer: show the substituted structures
v = WeasWidget()
v.from_ase(sColl.structures)
v.avr.model_style = 1
v.avr.color_type = "VESTA"
v.avr.boundary = [[-0.1, 1.1], [-0.1, 1.1], [-0.1, 1.1]]
v.avr.show_bonded_atoms = True
v
additionGen operates similarly, adding one or more atoms of the given species to a certain selection, and picking the direction to add them in a way that it will be as far as possible from existing bonds, again, under verification of an acceptance function if required.
# We try adding hydrogen to ethylene
eth = molecule('C2H4')
csel = AtomSelection.from_element(eth, 'C')
aG = additionGen(eth, 'H', to_addition=csel, add_r=1.0)
aColl = AtomsCollection(aG)
# Should generate four structures: 2 carbon atoms x 2 possible directions (above and below the plane of the molecule)
print('{0} structures generated'.format(len(aColl)))
4 structures generated
# Interactive viewer: compare H-addition structures
# The added H is appended at index len(eth), i.e. index 6 in each structure
from weas_widget import WeasWidget
added_h_idx = len(eth) # index of the newly added H atom
v = WeasWidget()
v.from_ase(aColl.structures)
v.avr.model_style = 1
v.avr.color_type = "VESTA"
v.avr.show_bonded_atoms = True
v.avr.highlight.settings['added_H'] = {
"indices": [added_h_idx],
"type": "sphere",
"color": [1.0, 0.4, 0.0],
"scale": 1.4,
"opacity": 0.5,
}
v.camera.setting = {"direction": [-1, -1, 0], "distance": 10, "zoom": 2}
v
2 - Analysing defect structures#
A different problem when dealing with the results of random structure searching is how to group together a number of predicted defect positions that might be crystallographically equivalent. In Soprano, this is helped by a clustering gene called defect_asymmetric_fdist, for “asymmetric fractional distance”. This is a pair gene (it only computes a distance between pairs of structures and only allows for hierarchical clustering), and it computes how close in fractional coordinates two defects can get if all possible symmetry operations that apply to a given pure structure are tried to bring them together. The distance itself has no specific physical meaning as it’s in fractional space, but it works well to group the defects together if there’s a meaningful structure to begin with. The gene requires having the space group library spglib installed.
from scipy.cluster.hierarchy import dendrogram
from soprano.analyse.phylogen import PhylogenCluster, Gene
# Let's create a number of different possible sites in silicon, split in two groups: tetrahedral and bond-centred site,
# but within a number of equivalent coordinates, and with some noise added in.
def_fpos = np.array([
[0.125]*3,
[0.125*5, 0.125, 0.125],
[0.125]*3,
[0.125*5, 0.125, 0.125],
[0.5]*3,
[0.75]*3,
[0.5]*3,
[0.75]*3
])
def_fpos += (np.random.random(def_fpos.shape)-0.5)*0.05
defColl = AtomsCollection([Atoms('H', scaled_positions=[fp], cell=si2.get_cell())+si2 for fp in def_fpos])
# Specify that the defect is at index 0, and that the pure structure is si2
dafGene = Gene('defect_asymmetric_fdist', params={'struct': si2, 'index': 0})
defClust = PhylogenCluster(defColl, genes=[dafGene])
# Interactive viewer: show the substituted structures
v = WeasWidget()
v.from_ase(defColl.structures)
v.avr.model_style = 1
v.avr.color_type = "VESTA"
v.avr.boundary = [[-0.1, 1.1], [-0.1, 1.1], [-0.1, 1.1]]
# Highlight the defect atom (index 0) with a sphere
v.avr.highlight.settings['defect'] = {
"indices": [0],
"type": "sphere",
"color": [0.2, 0.5, 0.9],
"scale": 3.4,
"opacity": 0.5,
}
v
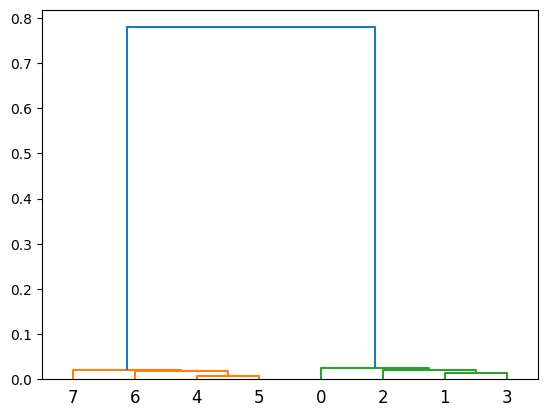
Z = defClust.get_linkage()
fig = plt.figure()
ax = fig.add_subplot(111)
# The structures should be neatly split in two clusters of four
dd = dendrogram(Z, ax=ax)